|
|
||||
|
Published by : PROFESSIONAL MEDICAL PUBLICATIONS |
||||
|
ISSN 1681-715X |
||||
|
||||
|
- |
||||
|
CASE SERIES |
||||
|
- |
||||
|
Volume 24 |
July - September 2008 |
Number 4 |
||
|
|
||||
|
|
||||
|
|
||||
|
Published by : PROFESSIONAL MEDICAL PUBLICATIONS |
||||
|
ISSN 1681-715X |
||||
|
||||
|
- |
||||
|
CASE SERIES |
||||
|
- |
||||
|
Volume 24 |
July - September 2008 |
Number 4 |
||
|
|
||||
|
|
||||
Turner’s syndrome: Diagnosis and
management with limited resourcesOgbera AO1, Onyekwere C2, Chinenyes S3
SUMMARY
Turner Syndrome or Bonnevie-Ulrich syndrome is a syndrome of gonadal dysgenesis characterized by sexual infantilism, short stature and somatic anomalies. These case reports are meant not only to describe the clinical features of Turners syndrome but also illustrate the challenges faced in the bid to diagnose this medical condition with the available limited diagnostic tools.
KEY WORDS: Turner’s Syndrome, Sexual Infantilism, Karyotype.
Pak J Med Sci July - September 2008 Vol. 24 No. 4 604-608
How to cite this article:
Ogbera AO, Onyekwere C, Chinenyes S. Turner’s syndrome: diagnosis and management with limited resources. Pak J Med Sci 2008;24(4):604-8.
1. Ogbera AO,
Department of Medicine,
2. Onyekwere C,
Department of Medicine,
1,2: Lagos State University Teaching Hospital,
Ikeja, Lagos, Nigeria.
3. Chinenyes S,
Department of Medicine,
University of Port-Harcourt Teaching Hospital,
Post-Harcourt, Nigeria.
Correspondence
Ogbera AO,
E-mail: oogbera@yahoo.co.uk
ifymobi@yahoo.com
* Received for Publication: October 8, 2007
* Revision Received: July 1, 2008
* Revision Accepted: July 11, 2008
INTRODUCTION
Turner’s syndrome a chromosomal disorder characterized by gonadal dysgenesis was first described in 1938. Other features of Turners syndrome include short stature and a variety of somatic anomalies.
1,2 The birth prevalence of Turner’s syndrome has been estimated to be from one in 2000 to one in 5000 female live births.2 Turner’s syndrome is not always accompanied by distinctive features and most often is not diagnosed in infancy. However later in childhood short stature may become obvious while in adulthood, the prominent features are those of sexual infantilism.1-4 The varied manifestations and the far reaching effects in the persons affected are also depicted in this case series.CASE REPORT
Case 1: The patient, a 17 year old girl presented with a history of delay in attainment of puberty –as at age 15 she was yet to attain secondary sexual characteristics and had to have sex hormone replacement therapy for a year from the referral centre. She also complained of inability to walk properly. (She walked with a limp). She had had a fall some few months prior to presentation and the fall was described as "mild" and out of proportion to the degree of the injury sustained.
Her referral letter showed biochemical evidence of hypothyroidism at initial presentation for which she had thyroxine replacement and later developed features clinically and biochemically of thyrotoxicosis. As at presentation, she was on carbimazole, propanolol and microgynon.
She is the only girl and the last child in a family of four children. She is presently in school and her academic performance is said to be below average compared to her siblings who are all said to be doing well academically. She had to repeat two classes.
On questioning, the mother said she had an uneventful pregnancy. Developmental milestones of the child in question were comparable to those of her siblings. There was no history of late menarche in the mother and her relations. There was a family history of Diabetes mellitus (DM)-the patient’s mother and paternal grandfather had DM. Drug history revealed that earlier she had thyroxine replacement for hypothyroidism and as at presentation was on carbimazole and propanolol. She also had microgynon prior to presentation to us. The review of the systems was unremarkable.
On general examination, she walked with a limp, otherwise she looked well. She had a high arched palate, a low hairline on the back, short 4
th metacarpal and puffiness of the dorsum of the fingers. Her neck was short and webbed. There was a firm non-tender goitre (WHO Grade 1B)5 whose size was estimated to be about 30g.Her weight was 69.3Kg while her height 1.52m, giving a BMI of 31Kgm
-2. The length of her upper body segment was 71cm while that of the lower body segment was 81cm. The ratio of the upper body segment to the lower body segment was 0.8. The mother’s height was 1.58 m, the father’s was 1.62m giving a mid parental height of 1.53 m using the formula.6 (For a girl), the approximate future height is [Paternal height–13cm + maternal height]/2Breast staging and Pubic hair staging according to the staging by Marshall and Tanner’s
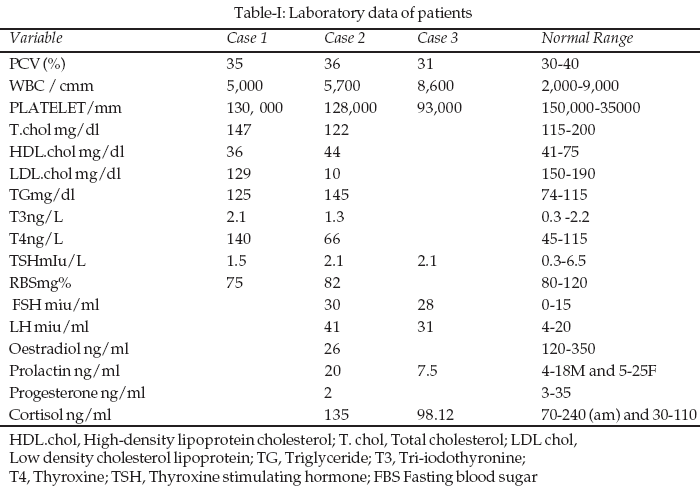
5 were Stages three and five respectively. She appeared clinically euthyroid and biochemical parameters as shown in Table-I which confirmed her euthyroid status.The cardiovascular system examination was essentially normal. On mental examination, she could not go beyond the first subtraction in the serial seven tests. Long term, short-term memories & immediate recall were unaffected. Abstract and judgement were impaired. The investigation results are as shown in Table-I.
Case 2: The patient, a twenty one years old girl was referred to us with a diagnosis of Gonadal dysgenesis. The essential features of the history included that of sexual infantilism and short stature. As at age seventeen years, she was yet to develop secondary sexual characteristics. She was noticed to be growing well up till when she was ten years old when it was noticed that her peers were growing taller than her. Further questioning revealed that though she was the first in a family of four children, her younger siblings were taller than her and way ahead of her academically. When she presented to us, she had just taken her school examinations with poor grades.
Drug history was essentially that of usage of conjugated equine estrogen 1.25mg daily which she was placed on at the age of 17 years following which she had some degree of breast and pubic hair development and actually started her menstrual cycles. She had no past history of trauma and there was no family history of delayed menarche. On examination, she was found to have short stature. Her anthropometric indices are as shown below: She weighed 48Kg while her height was 1.39m thus giving a BMI of 24.8 Kg m
-2. The proportion of her upper body segment to that of the lower body segment was 66 cm: 73cm giving a ratio of <1. Her arm span of 1.48m was greater than her height. Mid parental height using the Fathers height of 1.82cm and the mother’s height of 1.62m was 1.65m.Breast staging according to the staging by Marshall and Tanner’s
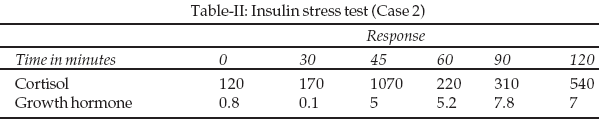
7 was stage two and Pubic hair stage three. Her cardiovascular system examination was essentially normal. She had biochemical evidence of hypogonadism. An insulin stress test revealed no abnormalities in cortisol and growth hormone response.Case 3:
An 18 year old girl presented with primary amenorrhoea and short stature. She was eight years old when it was noticed that she was not growing properly. She is the first in a family of three girls. Her siblings are all taller than her and had attained puberty. She is not known to have any chronic medical condition and she has no history of trauma. Her school grades have been good and comparable to her peers. There was no family history of sickle cell disease and delayed menarche. On examination, she was found to have short stature. Her anthropometric indices are as shown below:She weighed 34Kg while her height was 1.26m thus giving a BMI of 21.5Kg m
-2. The proportion of her upper body segment to that of the lower body segment was 58cm: 68cm giving a ratio of <1. Her arm span of 1.46m was greater than her height. Breast staging according to the staging by Marshall and Tanner’s 7 was stage one and Pubic hair stage two. Her pulse rate was 84 beats per minute, regular and was of a normal volume. Her cardiovascular system examination was essentially normal. Results are shown in Tables-I & II.
Radiological investigations:
Case 1: An abdominal ultrasound scan revealed no abnormality. A pelvic ultrasound scan showed an infantile uterus but no ovaries were visualized. X-ray showed dislocation of the hip joint, arthritic changes and features of longstanding slipped right femoral epiphysis with secondary femoral neck shortening. An X-ray of the left knee done for the determination of the bone age revealed a bone age of greater than 15 years. A buccal smear showed that 10% of the cells had Barr bodies.
Case 2: A pelvic ultrasound showed hypoplastic/ rudimentary uterus and streak gonads. X-ray of the left knee and left wrist showed delayed fusion of the epiphyses. An X-ray of the skull revealed no abnormality. Karyotype studies showed an XO chromosomal pattern
A Computed tomographic scan of the brain showed that the pituitary gland appeared small otherwise no focal brain lesions were detected. An intravenous urography showed bifid urinary collecting system. No abnormality was detected on echocardiography.
Case 3: A pelvic ultrasound showed an infantile uterine structure and streak gonads.
Buccal Smear: For Case number one this showed that 10% of the cells had Barr bodies while for Case two and Case three, Barr bodies were absent.
MANAGEMENT
For the three patients, their parents were educated on Turner syndrome and the potential complications. Available options in terms of achieving and maintaining secondary sexual characteristics, screening and prevention for potential complications and reproductive capacity were discussed.
Hormonal replacement with a combination of progesterone and Oestradiol were continued for both patients while for Case one, the dose of carbimazole was reduced to 5mg daily. Both patients are being seen on a follow up basis. The third patient was lost to follow-up.
DISCUSSION
Diagnosis in these cases posed some challenges in terms of laboratory work up since payment for tests were borne solely by the parents of these patients. Only one of the subjects was able to afford a karyotype study and this revealed an XO pattern while the other two had buccal smears taken to determine the presence of Barr bodies. Mosaic form of Turner’s was the diagnosis made in the subject who had a positive buccal smear (10% of the cells had Barr bodies) and some classical phenotypic features of Turners viz wide neck, shield like chest, wide carrying angle, low hair line high arched palate in addition to thyroidal and skeletal complications of this syndrome. All three subjects however had infantile uterine structures and absent to the presence streak gonads Asymptomatic renal abnormalities of the collecting duct (bifid collecting ducts) seen on intravenous urography was documented in the subject who were extensively investigated.
These case series have shown the varied manifestations of Turner’s syndrome. While the first and third cases had classical Turner’s syndrome as shown by the presence of an XO karyotype pattern and absence of Barr bodies in the buccal smears done, the second case was most likely, Mosaic form of Turner’s syndrome. Pointers to this diagnosis in the second case were essentially those of the clinical and in particular the laboratory features, A buccal smear showed the presence of Barr bodies in 10% of the cells in the buccal smear while the patient would not be considered as having short stature since the height was about that estimated for the mid-parental height. In the Mosaic form of Turners syndrome, Barr bodies are positive in 3-19% of the buccal smear cells.
8,9One clinical feature that was in keeping with the suspected diagnosis of Mosaic Turners is the height of the patient. Her height of 1.52m was close to that of the mid-parental height which was 1.53m. This is far above the estimated final height of most patients with Turners syndrome which is usually given as being between 1.42-1.47m.
10,11 However she had features of typical Turners viz a broad shield like chest, webbed neck, high arched palate and puffiness of the dorsum of the fingers12,13 just to mention a few. Apart from short stature-with a height of 1.39cm way below the estimated mid-parental height of 1.65m. The second patient in whom the definitive diagnosis of "Classical" Turners was made did not have the obvious phenotypic features consistent with this syndrome. For the third patient, the diagnosis was straight forward given the absence of Buccal smear and the clinical features of short stature and hypogonadism.Gonadal dysgenesis which is an invariant feature of people with Turners syndrome was seen in all three patients. All except one that was lost to follow-up had to have sex hormonal replacement to be able to achieve some degree of sexual development. The presence of sexual infantility was what prompted the parents of these patients to seek medical attention. That the prospect of infertility is by far the most existential threat to patients with Turner’s
6 has been exemplified in this report where the parents of these patients were worried about their reproductive capacity. Medical help was sought in all three cases long after abnormal growth pattern was noted.Somatic complications of Turners were noted in the first two cases. The patient in case one had thyroid abnormalities which alternated between thyrotoxicosis and hypothyroidism. This made us to suspect the presence of Hashimotos disease, which is an auto immune disorder that is reported to be more prevalent in people with Turner syndrome than in the general population.
14 This same patient also manifested some of the skeletal abnormalities seen in people with Turners syndrome viz arthritis and pathological fracture from osteoporosis. The arthritis may have resulted from congenital developmental dysplasia of the hip joint – a disorder seen commonly in Turners syndrome and contributing to the development of arthritis in such patients.15 The patient in case two had an abnormality in the urinary tract-bifid collecting tract- detected only when an intravenous urography was undertaken as part of routine screening for somatic anomalies of Turners syndrome. Urinary tract abnormalities are said to occur in one third of patients with Turners and these abnormalities may pretend an increased risk of hypertension and urinary tract infections14 thus underscoring the need for routine screening for these complications.One notable feature of both patients is that though not in any way mentally retarded, their academic performance fell way below that of their siblings. This is not surprising as learning difficulties are often reported in these groups of patients.
The management of Turner’s is usually multidisciplinary and each case is treated on its own merit. Available treatment options include sex hormonal replacement for hypogonadism and growth hormone for correction of short stature. Sex hormonal replacement is also administered because of its ability to prevent osteoporosis. Our patients did not have growth hormonal replacement because they presented late and also because it is not available in our practice. It is hoped that this report will sensitize clinicians to the elaborate features of Turners so that prompt diagnosis and management especially that geared towards correcting short stature and preventing osteoporotic complications are made early.
REFERENCES
1. Massa GG, Vanderschueren–Lodeweyckx M. Age and height at diagnosis in Turner syndrome: Influencer of parental height. Paediatrics 1991;1148-52.
2. Savendahl L, Davenport ML. Delayed diagnosis of Turners syndrome: proposed guidelines for change. J Paediatrics 2000;137:455-9.
3. Hsu LYF. Prenatal diagnosis of chromosomal abnormalities through amniocentesis. In: Milunsky A ed. Genetic disorders and the fetus. Baltimore: The John hopkins University Press, 1998;179-248.
4. Robinson A. Demography and Prevalence of Turner’s syndrome In: Rosenfeld RG, Grumbach MM, eds. Turner’s Syndrome. New York: Marcel Dekker 1990;93-9.
5. Rojeski MT, Gharib H. Nodular thyroid disease. New Eng J Med 1985;313:428-35.
6. Van Wyk JJ, Under wood LE. Normal and Aberrant growth. In: Williams and Foster editors. William’s Textbook of Endocrinology. 8th ed. Dallas. Saunders: 1992;1106-10.
7. Tanner JM. Growth at adolescence. 2nd ed. Oxford: Blackwell Scientific Publications 1962.
8. Soenger P, Ranke MB. Turner’s syndrome. The Lancet 2001;358:309-13.
9. Barr ML, Bertram EG. A morphological distinction between neurones of the male and the female and the behaviour of the nucleolar satellite during acceleration of nucleoprotein synthesis. Nature 1949;163:676-7.
10. Tarani L, Lampertello S, Raguso G. Pregnancy in patients with Turner’s syndrome-a psychosocial report from 22 middle-aged women. Acta Endocrinol 1998;12:83-7.
11. Sylven L, Magnusson C, hagenfeldt K, Von Schultz B. Life with Turner’s Syndrome:six new cases and review of literature. Gynecol Endocrinol 1998;12:83-7.
12. Pasquino AM, Pucarelli I, Segni M. Spontaneous puberty in Turner’s syndrome. IN: Saenger P, Pasquino AM eds. Optimizing health care for Turner’s patients in the 21st century. Elsevier, 2000;231-8.
13. Williams JK. School aged children with Turner’s Syndrome. J Paediatr Nurs 1992;7:14-19.
14. Elsheikh M, Conway GS, Wass JA. Medical problems in children with Turner’s syndrome. Annals of Internal Medicine 1999;31:99-105.
15. Saenger P, Albertsson-Wikland K, Conway GS. Fifth International symposium on Turner syndrome. Recommendations for the diagnosis and management of Turner syndrome. J Clin Endocrin Metabolism 2001;86:3061-9.
HOME | SEARCH | CURRENT ISSUE | PAST ISSUES
Professional
Medical Publications
Room No. 522, 5th Floor, Panorama Centre
Building No. 2, P.O. Box 8766, Saddar, Karachi - Pakistan.
Phones : 5688791, 5689285 Fax : 5689860
pjms@pjms.com.pk