|
|
||||
|
Published by : PROFESSIONAL MEDICAL PUBLICATIONS |
||||
|
ISSN 1681-715X |
||||
|
||||
|
- |
||||
|
ORIGINAL ARTICLE |
||||
|
- |
||||
|
Volume 24 |
April - June 2008 (Part-II) |
Number 3 |
||
|
|
||||
|
|
||||
|
|
||||
|
Published by : PROFESSIONAL MEDICAL PUBLICATIONS |
||||
|
ISSN 1681-715X |
||||
|
||||
|
- |
||||
|
ORIGINAL ARTICLE |
||||
|
- |
||||
|
Volume 24 |
April - June 2008 (Part-II) |
Number 3 |
||
|
|
||||
|
|
||||
Spectrum of ▀ ¢Thalassemia mutations
in various Ethnic Regions of Iran
Rahim F1, Abromand M2
ABSTRACT
Objective: Thalassemia is a group of inherited disorders characterized by reduced or absent amounts of hemoglobin, the oxygen-carrying protein inside the red blood cells. ▀-Thalassemia, one of the most widespread genetic disease in the world, is a common autosomal recessive disorder generally caused by point mutations in the ▀-globin gene that is located as a cluster on the short arm of chromosome 11(OMIM:MIM # 141900). The objective of this study was to identify spectrum of Beta Thalassemia mutations in various ethnic regions of Iran.
Methodology: We extracted and differentiated the Iron deficiency patients with the help of 10 discrimination indices (Mentzer Index, England and Fraser Index, Srivastava Index, Green and King Index, Shine and Lal Index, red blood cell (RBC) count, red blood cell distribution width, red blood cell distribution width index (RDWI), Mean Density of Hemoglobin per Liter of blood (MDHL) and Mean Cell Hemoglobin Density (MCHD)) from beta-thalassemia patients.
Results: In a total of 1098 carriers (1045 beta-thalassemia and 53 iron deficiency), we detected different ▀-thalassemia mutations in the studied subjects of 5 different ethnic regions from Iran. ▀-Thalassemia was diagnosed based on complete blood counts, Hb electrophoresis, and ASO-Hybridization in patients from each area separately at Research center of Thalassemia and Hemoglobinopathies, Ahwaz Jondishapur University of Medical Sciences, Shafa Hospital, Iran. This study has shown that the most common mutation for each region was IVS-II-1 (G ăåÆ A) (34%) in Khuzestan, IVS I (3' end)-25 bp (28.7%) in Booshehr, IVS II- 1(G ăåÆ A) (41.5%) in Fars, IVS-II-1 (GăåÆ A) (31.8%) in Isfahan, IVS I-5 (GăåÆ C)(44.8%) in Sistan- Baloochestan, respectively.
Conclusions: The presence of such a high frequency of various local mutants alleles is strong support for role of non-isolating genetically areas. In likelihood, both founder effect and natural selection caused by migration from neighboring areas have complemented each other to produce the high frequency of unique alleles within each region.
KEYWORDS: ▀-Thalassemia, Iron deficiency, Differential indices, ▀-globin gene, IVS (intervening sequence), CD (Codon), OMIM.
Pak J Med Sci April - June 2008 (Part-II) Vol. 24 No. 3 410-415
How to cite this article:
Rahim F, Abromand M. Spectrum of ▀ ¢Thalassemia mutations in various Ethnic Regions of Iran. Pak J Med Sci 2008;24(3):410-5.
1. Fakher Rahim,
Research Center of Thalassemia and Hemoglobinopathies,
2. Muhammad Abromand,
Department of Biochemistry and Genetics,
1-2: Ahwaz Jondishapur University of Medical Sciences,
Iran.
Correspondence
Fakher Rahim,
E-mail: Fakherraheem@yahoo.com
* Received for publication: January 17, 2008
* Revision Received: March 24, 2008
* Revision Accepted: March 25, 2008
INTRODUCTION
The thalassemias are widespread with about 5% of the world population affected by it. It is most prevalent around the Mediterranean Sea i.e. countries like Greece, Italy, Turkey and North African countries. It is also seen in Saudi Arabia, Iran, Afghanistan, Pakistan India and South East Asian countries like Thailand and Indonesia. Iran, a country spread over 1,648,000km two wide, has a large number of thalassemia major patients like many other countries in the region.
1 ▀-thalassemia is very rare in Iran. The gene frequency of ▀-thalassemia, however, is high and varies considerably from area to area, having its highest rate of more than 10% around the Caspian Sea and Persian Gulf. The prevalence of the disorder in other areas is between 4-8%. In Isfahan, a city built around the river Zayandeh-Rood in the central part of Iran, the frequency rises again to about 8%. In the Fars Province, in southern Iran, the gene frequency is also high and reaches 8-10%.1Beta-Thalassemia, one of the most widespread genetic disease in the world, is a common autosomal recessive disorder generally caused by point mutations in the ▀- globin gene that is located as a cluster on the short arm of chromosome 11.
2-4 More than 200 different mutations affecting diverse levels of beta-globin genes expression have so far been identified.3,5,6 Different strategies of classification individuals genotypes by ▀-globin gene cluster and cloning nucleotides sequencing lead to identification of several mutations in Mediterranean,7 Asian Indians,8,9 American Blacks10 and Chinese.11 The global distribution indicates a high prevalence in a belt around the earth, which is around the 40th parallel in the Mediterranean area but eastwards moves further south, reaching the equator in Indonesia.More than two million carriers of ▀-thalassemia live in Iran. Since the Iranian populations are mixture of different ethnic groups, it is necessary to determine the frequency and distribution of mutations in the different parts of the country. For this purpose, we studied ▀-thalassemia chromosomes of total 1098 affected patients (1045 beta-thalassemia and 53 iron deficiency) by using the ASO-Hybridization. We detected frequency of different ▀-thalassemia mutations in the studied subjects of five different ethnic regions from Iran including Isfahan, Sistan- Balochestan, Khuzestan, Booshehr and Fars.
Methodology
▀-Thalassemia was diagnosed based on complete blood counts, Hb electrophoresis, and ASO-Hybridization of total 1045 beta-thalassemia carriers at Research Center of Thalassemia and Hemoglobinopathies, Ahwaz Jondishapur University of Medical Sciences, Shafa Hospital, Iran. The hematological data indicates the low MCV (Mean Corpuscular Volume) and low MCH (Mean Corpuscular Hemoglobin) values and high or borderline Hb A2 fraction in heterozygous state. Due to the prevalence of thalassemia in Iran, a descriptive cross-sectional study was conducted to determine a more valid variable for screening minor thalassemia patients.
After initial abnormal blood count as described above, since iron deficiency is the other explanation for low MCV or MCH, we used different indices for differential diagnosis between these two disorders and found the Mean and Standard deviation of hematological values as shown in Table-I. We calculated 10 discrimination indices such as Mentzer Index,
12 England and Fraser Index,13,14 Srivastava Index,15 Green and King Index,16 Shine and Lal Index,15 Red blood cell (RBC) count, red blood cell distribution width and red blood cell blood distribution width index (RDWI),17 MCHD Index,18 MDHL Index.19 Other two indices include Red blood cell count (RBC) and red blood cell distribution widths (RDW)20,21 were obtained with Counter. We could differentiate the beta-thalassemia patients (1045) from iron deficiency (53) with the help of indices results successfully in total 1098 subjects.DNA Isolation:
5ml Peripheral blood samples were collected in tubes containing EDTA. We used Viena Lab Kit.DNA Extraction:
DNA was extracted from the blood sample cells by using the Viena Lab Kit. This Kit contains two parts i.e. Lyris Solution and GENXTRACT Resin. The DNA extraction procedure was: (i) put 100Ąl blood sample in micro tube, add 1ml Lyris Solution, incubate 15 minutes in room temperature, spin down at 3000 rpm for 5 minutes, (ii) add 1ml Lyris Solution to extracted supernatant, incubate 5 minutes in room temperature, spin down at 12000 rpm for 10 minutes. (iii) add 200Ąl GenXtract to supernatant, incubate 20 minutes at 56║C (iiV) incubate 10 minutes at 98║C and spin down at 3000 rpm fr 5 minutes. Finally, we extracted supernatant that involves pure DNA and use it in next step.PCR Amplification:
In vitro amplification of genomic DNA was performed with HotStarTaq Master Mix Kit Qiagen by PCR technique in Gene Amp PCR System 2400 thermal-cycler (Mastercycler 5330), using different sets of primers including whole structural and untranslated regions of ▀-globin gene, described by Kanavakis et al.22 The amplification cycle consisted of: An initial denaturation at 95║C for 15 min was followed by 35 cycles of a three-step cycling protocol (95░C for 1 minutes, 60░C for 1 minutes, and 72░C for 90 seconds) and a final elongation step at 72░C for 8 min. PCR reaction composition using HotStarTaq Master Mix were prepared in mixtures of 2.5units HotStarTaq DNA Polymerase, 1ūPCR buffer, 200 mm of each dNTP, 0.1¢0.5mm of each primer, 1ml Template DNA.Hybridization:
Hybridization of PCR products was done using different solutions including DNAT (1.6% NaOH) and Hybridization Buffer by mix 10Ąl of DNAT with 10Ąl of PCR product, add 1ml Hybridization Buffer at 45║C and keep it on shaker for 5 minutes.Washing:
After 30 minutes washing process was started with Wash solution A, three times each time for 15 minutes at 45 ║C on shaker.Chromatography:
Chromatography was applied to the bands using solutions including Wash solution B, Conjugation Solution, and Color Developer. Method applied by adding 1ml Conjugation Solution to the samples, removes the Conjugation Solution after 15 minutes and adds 1ml of Wash solution B twice. Finally apply 1ml of Color Developer solution for 15 minutes in dark place.RESULTS
Analysis of Mutation:
This study was initiated by screening genomic DNA sample of the patients in order to find different mutations and concluded the development of PCR follows by hybridization. Blood samples were collected from 1045 patients (260 less than 10 years and 785 more than 10 years) of five different areas of Iran. DNA was isolated and the samples were subjected to gene amplification. The primers were selected on the basis of location. The amplified DNA samples were screened for the presence or absence of different mutations. Denatured DNA samples were spotted onto nylon membranes and hybridized to oligonucleotide probes for frame-shift mutation in corresponding sequences. At the next stage, DNA samples carrying unknown alleles were screened for common mutations.
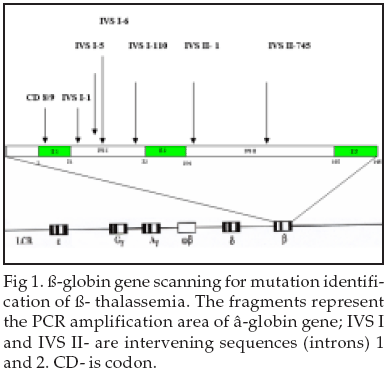
▀- Globin Mutations: In our study several known thalassemia mutations with different frequency in various areas has been found (Fig-1). The most common known mutations with higher frequency were detected in each ethnic region of interest. Results have shown that most frequent mutation includes Mediterranean7,23-25 Asian, Indian and Japanesem8,9 and SE Asian, Melanesian.26,27 We found that the IVS II- 1(G ăåÆ A) mutation was the most common ▀-thalassemia defect with frequency of 26.5% (277/1045), followed by IVS I-I (G ăåÆA) (12.8%), CD 8/9 (12.3%), IVS I-5 (11.2%), IVS I - 110 (11%) among all five provinces (Table-I).

We also calculated the related frequency for each area separately (Table-I). The most common mutation for each region was IVS-II-1 (G ăåÆ A) (34%) in Khuzestan, IVS I (3' end)-25 bp (28.7%) in Booshehr, IVS II- 1(G ăåÆ A) (41.5%) in Fars, IVS-II-1 (G ăåÆ A) (31.8%) in Isfahan, IVS I-5 (G ăåÆ C)(44.8%) in Sistan- Baloochestan, respectively (Table-I).
DISCUSSION
Iran is a country which has a population with a different ethnic identity and different languages. The people who live in different parts speak different languages, but the common language is Persian. As we saw mutation in ▀-globin gene will lead to thalassemia. Although, all this mutations were found in coastline areas but their geographic distribution has special characteristic properties. In most parts of the world, a small number of thalassemia mutations predominant and the most common ones tend to be those that are geographically the most widespread and presumably also the oldest. For instance, in China and Southeast Asia, four alleles account for 91% of the genes
30 and in the Mediterranean Basin, six mutation account for 92% of the genes.31 However, Mutation 619 bp deletion is predominant in India or mutations IVS I-110 & IVS II-1 are most predominant in Arab populations.28 Because of Wars and trade off between different countries in the past years there was exchange of genetic materials between different populations.We attempted to detect the most frequent ▀-thalassemia mutations in Khuzestan and Booshehr (Southwest and Southeast regions), Isfahan and Fars (Central Part). Different authors have reported various mutations that are most prevalence in Khuzestan. In a work done by Karimi et al., on 87 patients with ▀- thalassemia intermedia using ARMS technique, they have detected four different mutations involves IVS II-1, IVS i-110, IVS I-1, and CD 8/9. They claimed that these mutations are the most frequent in Iran and IVS II-1(24%) is most predominant in Khuzestan.
29 In a similar work on 1217 patients with ▀- thalassemia the most predominant mutation in North part of Iran was IVS II-1(34%) and in South part was IVS I-5(%).30Najmabadi et al. have studied ▀ -globin mutations and claimed that the most common ▀ -globin mutations is IVS I-130 (G-C), which was identified in six subjects from the North of Iran, three subjects from the Southwest, as well as in one DNA of unknown geographical origin.
31 Karimi et al. have reported the highest prevalence mutations of IVS-II-1, IVS-I-110, IVS-I-1 and CD 8/9. They claimed that the IVS-II-1 defect, being the most frequent in south of Iran, was present at the highest rate (24%).32 We have found that most predominant in South was IVSII-I (34%) follows by IVS I-110(15.7%) and CD 8/9(13.3%). Previous research work showed most predominant mutation in Pakistan33 is IVS I-5(37%) therefore this mutation is most predominant in Sistan-Baloochestan (44.8%)(Southeast of Iran) because it is a neighborhood area to Pakistan. In a work done on thalassemia patients in Hormozgan (Southern part of Iran) claimed that most predominant mutation there is IVS I-5(69%) followed by IVS II- 1(9.6%).33Research work done in 8 Gulf (Arab) Countries showed that most predominant mutations are IVS I-110 and IVS II-1 followed by IVS I-5, CD39, CD6, IVS I(3' end)-25 bp del.
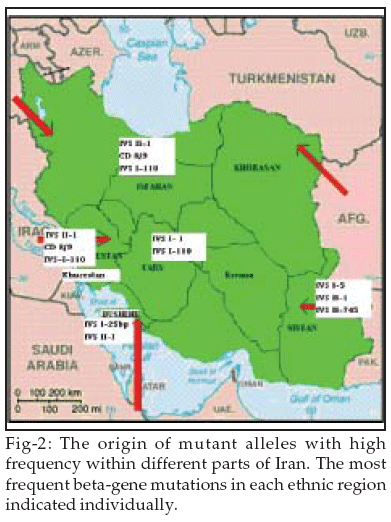
34 Our finding was similar to the work done in Kuwait which showed 6 mutations are most predominant involve IVS II-1, IVS I-6, CD39, IVS I-110, CD8, IVS I-1(all give 64%) and followed by another 2 involve CD44, CD 36/37 (Kurd, Iranian types) that give 10% of the population.35 We detected different ▀-thalassemia mutations in the studied chromosomes and 5 different areas from Iran which showed IVS-II-I (G ăåÆ A) was the predominant mutation found in all ethnic regions. The most common mutation for each region was IVS-II-1 (G ăåÆ A) (34%) in Khuzestan, IVS I (3' end)-25 bp (28.7%) in Booshehr, IVS II- 1(G ăåÆ A) (41.5%) in Fars, IVS-II-1 (G ăåÆ A) (31.8%) in Isfahan, IVS I-5 (G ăåÆ C)(44.8%) in Sistan- Baloochestan, respectively(Table-I). The presence of such a high frequency of various local mutants alleles confirms support for a role of non-isolating genetically areas. In likelihood, both founder effect and natural selection caused by migration from neighboring areas have complemented each other to produce the high frequency of unique alleles within each region (Fig-2). The results presented here can be used as a basis of prenatal diagnosis of ▀-thalassemia.ACKNOWLEDGMENTS
The authors are extremely grateful to all staffs of Research Center of Thalassemia and Hemoglobinopaties, Ahwaz Jondishapour University of medical sciences, Shafa hospital in Iran for their help and co-operation.
REFERENCES
1. Haghshenas M, Zamani J. [Thalassemia]. 1st ed. Shiraz University of Medical Sciences Publishing Center: Shiraz; 1997. [Book in Persian]
2. Weatherall DJ, Clegg JB, Higgs DR, Wood WG. The hemoglobinopathies. The Metabolic and Molecular Bases of Inherited Disease. McGraw - Hill, New York. 1989;3417-83.
3. Sack GH. Autosomal recessive disorders. Medical Genetics, McGraw-Hill, New York. 1999;61-3.
4. Oliveri NF. The beta- thalassemias. New Eng J Med 1999;341:99-109.
5. Trent RJ. Medical Genetics. Molecular Medicin, Longman Singapore Ltd., Singapore. 1997;37-717.
6. Ho PJ, Thein SL. Gene regulation and deregulation: a beta globin perspective. Blood Rev 2000;14(2):78 -93.
7. Orkin SH, Kazazian HH, Jr Antonarakis SE, Goff SC, Boehm CD, Sexton JP, et al. Linkage of beta - thalassemia mutations and beta-globin gene polymorphisms with DNA polymorphisms in human beta-globin gene cluster. Nature 1982;296(5858):627-31.
8. Kazazian HH, Jr, Orkin SH, Antonarakis SE, Sexton JP, Boehm CD, Goff SC, et al. Molecular characterization of seven beta - Thalassemia mutations in Asian Indians. EMBO J 1984;3(3):593-6.
9. Ohba Y, Hattori Y, Harano K, Fukumaki Y, Ideguchi H. ▀-thalassemia mutations in Japanese and Koreans. Hemoglobin 1997;21:191-200.
10. Antonarakis SE, Irkin SH, Cheng TC, Scott AF, Sexton JP, Trusko SP, et al. beta - Thalassemia in American Blacks: novel mutations in the "TATA" box and an acceptor splice site. Proc Natl Acad Sci USA. 1984;81(4):1154-8.
11. Cheng TC, Orkin SH, Antonarakis SE, Potter MJ, Sexton JP, Markham AF, et al. beta- Thalassemia in Chinese: use of in vivo RNA analysis and oligonucleotide hybridization in systematic characterization of molecular defects. Proc Natl Acad Sci USA 1984;81(9):2821-5.
12. Mentzer WC. Differentiation of iron deficiency from thalassemia trait. Lancet 1973;1:882.
13. England JM, Fraser P. Discrimination between iron-deficiency and heterozygous-thalassemia syndromes in differential diagnosis of microcytosis. Lancet 1979;1:145-8.
14. England JM, Fraser PM. Differentiation of iron deficiency from thalassemia trait by Routine blood- count. Lancet 1973;1:449- 52.
15. Shine I, Lal S. A strategy to detect ▀-thalassemia minor. Lancet 1977;1:692- 4.
16. Srivastava PC, Bevington JM. Iron deficiency and-or thalassemia trait. Lancet 1973;1:832.
17. Green R, King R. A new red blood cell discriminant incorporating volume dispersion for differentiating iron deficiency anemia from thalassemia minor. Blood Cells 1989;15:481-95.
18. Jayabose S, Giavanelli J, Levendoglu-Tugal O, Sandoval C, Ozkaynak F, Visintainer P. Differentiating iron deficiency anemia from Thalassemia minor by using an RDW-based index. J Pediatr Hematol 1999;21:314.
19. TelmissaniI O A, Khalil S, TR George. Mean Density of Hemoglobin Per Liter of Blood: A New Hematologic Parameter With an Inherent Discriminant Function. Lab Haematol 1999;149-52.
20. Klee GG, Fairbanks VF, Pierre RV, Virgh D, OÆSullivan MB. Routine erythrocyte measurements in diagnosis of iron-deficiency anemia and thalassemia minor. Am J Clin Pathol 1976;66:870-7.
21. Bessman JD, Feinstein DI. Quantitative anisocytosis as a discriminant between iron deficiencies and thalassemia minor. Blood 1979; 53: 288-93.
22. Kanavakis E, Traeger-Synodinos J, Vrerrou C, Maragoudaki E, Tzetis M, Kattamis C, Prenatal diagnosis of thalassemia syndromes by rapid DNA analytical methods, Molec Hum Reprod 1997;3:523.
23. Spritz RA, Jagadeeswaran P, Choudary PV, Biro PA, Elder JT, DeRiel JK, et al. Base substitution in an intervening sequence of a ▀+ Thalassemia human globin gene. Proc Natl Acad Sci USA 1981;78:2455.
24. Westaway D, Williamson R. An intron nucleotide sequence variant in a cloned ▀+ Thalassemia globin gene. Nucleic Acids Research 1981;9:1777-88.
25. Tamagnini GP, Lopes MC, Castanheira ME, Wainscoat JS, Wood WG. ▀+ Thalassemia -Portuguese type: clinical, hematological and molecular studies of a newly defined form of beta- thalassemia. British J Hematology 1983;54:189-200.
26. TreismanR, Orkin SH, Maniatis T. Specific transcription and RNA splicing defects in five cloned beta- thalassemia genes. Nature 1983;302:591-6.
27. Hill AVS, Bowden DK, O┤Shaughnessy DF, Weatherall DJ, Clegg JB. Beta-thalassemia in Melanesia: association with malaria and characterization of a common variant. Blood 1988;72:9.
28. Nozari G, Rahbar S, Golshaiyzan A, Rahmanzadeh S. Molecular analysis of ▀- thalassemia in Iran. Hemoglobin 1995;19:425-31.
29. Orkin SH, Sexton JP, Goff SC, Kazazian HHJ. Inactivation of an acceptor RNA splice site by a short deletion in Beta-thalassemia. Bio Chem J 1983;258:7249.
30. Kazazian HH, Jr., Dowling CH, Waber PG, Hung S, LO, WHY. The spectrum of Beta - thalassemia genes in China and Southeast Asia. Blood 1986;68:964-6.
31. Najmabadi H, Ali A. Pourfathollah, Neishabury M, Sahebjam F. Rare and unexpected mutations among Iranian ▀ -thalassemia patients and prenatal samples discovered by reverse-hybridization and DNA sequencing. Haematologica 2002;87(10).
32. Karimi M, Yarmohammadi H, Farjadian S, Zeinali S, Moghaddam Z, Cappellini MD, et al. Beta-thalassemia intermedia from southern Iran: IVS-II-1 (GŚ>A) is the prevalent thalassemia intermedia allele. Hemoglobin 2002;26(2):147-54.
33. Kazazian HH. Jr., Orkin SH, Markham AF, Chapman CR, Youssoufian H, Waber PG. On the origin and spread of beta-thalassemia: recurrent observation of four mutations in different ethnic groups. Nature 1984;310:152-4.
34. El-Hazmi MA, Warsy AS, Al-Swailem AR. The frequency of 14 beta- thalassemia mutations in the Arab populations. Hemoglobin 1995;19(6):353-60.
35. Adekile A, Haider M, Kutlar F. Mutations Associated with Beta-Thalassemia intermedia in Kuwait. Medical Principles and Practice 2005;14(Suppl.1):69-72.
36. Hardison RC, Chui DHK, Giardine B, Rimer K, Patrinos GP, Anagnou N, et al. A Relational database of Human Hemoglobin Variants and Thalassemia Mutations at the Globin Gene Server. Human Mutation 2002;19:225-33.
HOME | SEARCH | CURRENT ISSUE | PAST ISSUES
Professional
Medical Publications
Room No. 522, 5th Floor, Panorama Centre
Building No. 2, P.O. Box 8766, Saddar, Karachi - Pakistan.
Phones : 5688791, 5689285 Fax : 5689860
pjms@pjms.com.pk