|
|
||||
|
Published by : PROFESSIONAL MEDICAL PUBLICATIONS |
||||
|
ISSN 1681-715X |
||||
|
||||
|
- |
||||
|
ORIGINAL ARTICLE |
||||
|
- |
||||
|
Volume 23 |
October - December 2007 (Part-I) |
Number 5 |
||
|
|
||||
|
||||
|
|
||||
|
Published by : PROFESSIONAL MEDICAL PUBLICATIONS |
||||
|
ISSN 1681-715X |
||||
|
||||
|
- |
||||
|
ORIGINAL ARTICLE |
||||
|
- |
||||
|
Volume 23 |
October - December 2007 (Part-I) |
Number 5 |
||
|
|
||||
|
||||
The Mitochondrial K-ATP Channel Opener, Diazoxide,
protects brain against Ischemia-Reperfusion Injury in the Rat
Mehdi Mehdizadeh1, Mansooreh Soleimani2,
Majid Katebi3, Hamid Reza Pazouki4,
Mohamad Taghi Joghataei5, Noor Ahmad Latifi6, Masoomeh
Bakhshayesh7
ABSTRACT
Objective: Even today there is no effective drug therapy to prevent neuronal loss after brain stroke. The objective of this research was to study effects of the mitochondrial K-ATP (MAK) channel regulators on neuronal cell population and neurological function after ischemia reperfusion in the rat.
Methodology: Rats were temporarily subjected to four vessels occlusion for 15 minutes followed by 24 hours reperfusion with or without MAK channel regulators.
Results: The normal cell count of neuronal population significantly increased in the K-ATP channel opener (diazoxide) treated ischemia-reperfusion group compared with the control group. Cell count and neurological function scores were dose dependent to MAK channel regulators in vivo.
Conclusions: Our results showed that diazoxide treatment leads to better preservation of cortical neurons in rat.
KEYWORDS
: K-ATP Channel, Ischemia reperfusion Injury, Diazoxide.Pak J Med Sci October - December 2007 (Part-I) Vol. 23 No. 5 741-746
1. Mehdi Mehdizadeh
2. Mansooreh Soleimani
3. Majid Katebi
4. Hamid Reza Pazouki
5. Mohamad Taghi Joghataei
6. Noor Ahmad Latifi
7. Masoomeh Bakhshayesh
1,2,5: Department of Anatomy,
School of Medicine,
3: Department of Medicine,
School of Medicine, New York University,
New York , NY , USA
4: Department of Physiology,
School of Medicine,
6: Department of Plastic Surgery
School of Medicine,
7: Cellular and Molecular Research Center
1,2,4-7: Iran University of Medical Sciences,
Tehran – Iran.
Correspondence
Mehdi Mehdizadeh,
Dept. of Anatomy, School of Medicine ,
Iran University of Medical Sciences,
Tehran – Iran.
E-mail: maranaoo2004@yahoo.com
* Received for Publication: April 30, 2007
* Revision Received: May 15, 2007
* Revision Accepted: August 30, 2007
INTRODUCTION
Stroke is a major cause of death in the developed countries.
1,2 Stroke has been considered as untreatable and even today there is no effective drug therapy to help stroke patients. There is increasing evidence that the functional recovery after cerebral lesions and ischemia may be influenced by pharmacotherapy.3 There is escalating evidence that mitochondria play a key role in both necrotic and apoptotic neuronal cell death after acute cerebral ischemia.4,5 Mitochondrial dysfunction is one factor that plays a critical role in mediating both apoptotic and necrotic neuronal cell death and is involved in the pathophysiology of cerebral ischemia.6-9 MAK channels have been distinct population of channels.10 There is increasing evidence about the diverse functions of these channels in the regulation of mitochondrial matrix volume and ATP production9 and represent a pharmacologically n, and Ca2 + homeostasis in mitochondria. These are essential factors determining the outcome of ischemic stress on cellular function and survival.7-10 It has been reported that MAK channel openers reduce mitochondrial Ca2+ overloading during ischemia reperfusion in isolated rat hearts.11 This may be a trigger to prevent an apoptotic signaling pathway through the mitochondria.There are no absolutely selective pharmacological tools to assess the MAK channels in vivo. However, a consistent and unique feature of these channels is their remarkably selective sensitivity to opening by diazoxide.
12 Many substances, including adenosine, acetylcholine, & opioids, protect against ischemia-reperfusion injury via opening of the channel.13-15 The mechanism by which activation of MAK channels protect ischemia remains to be clarified.MATERIALS AND METHODS
Animal Treatment: Wistar male rats weighing 180 to 200 g were used in this study. Rats were housed in an air-conditioned room with a 12-hour light/dark cycle, received a standard rat chow (0.4% sodium chloride), and drank tap water. All procedures complied with the standards for the care and use of animal subjects as stated in the "Guide for the Care and Use of Laboratory Animals" (Institute of Laboratory Resources, National Academy of Sciences). Rats were randomly assigned to the following groups: Group 1: animals were subjected to the anesthetic and surgical procedures of 4VO without interruption of the cerebral blood flow (Sham, n=6) ; "the third group was subjected to 4VO received glibenclamide, 1mg/kg (Experimental 1, n=6), and the fourth group was subjected to 4VO received glibenclamide, 5mg/kg (Experimental 2, n=6), and fifth groups was subjected to 4VO received glibenclamide, 25mg/kg (Experimental 3, n=6), and the sixth group was subjected to 4VO received diazoxide, 2mg/kg (Experimental 4, n=6), and seventh group was subjected to 4VO received diazoxide, 6mg/kg (Experimental 5, n=6), and eighth group was subjected to 4VO received diazoxide, 18mg/kg (Experimental 6, n=6).
Anesthesia and Monitoring:
Overnight animals were fasted anesthetized with ketamine (100mg/kg, IP) and Xylozine (10mg/kg IP) with additional ketamine as needed (30mg/kg, IP). A rectal temperature probe was placed for continuous monitoring of core body temperature. Core body temperature was maintained at 37±0.5°C throughout the procedure by the intermittent use of a heating lamp.Surgical Procedure:
Permanent vertebral artery (VA) occlusion and 15 minutes common carotid artery (CCA) occlusion was induced. A 1-cm dorsal midline incision was made in a sterile fashion at the occiput-C2 level to separate the Para spinal muscles from the midline. The alar foramina of C1 was isolated with the aid of an operating microscope, the VA’s cauterized with a 0.5mm electrocaurtery needle through the alar foramina, and the incision closed with 4-0 nylon. A second 1-cm midline incision was made in a sterile fashion over the ventral aspect of the neck, the CCA’s isolated, and a traumatic vascular occlusion clips placed onto each CCA. CCA occlusion was induced for 15 minutes after which the clip was removed. The incision was closed with 4-0 nylon and the animals were allowed to reperfusing and recover for 24 hours.Neurological Evaluation: Neurologic assessment was conducted after 24hour reperfusion by an observer unaware of the treatment groups. Consciousness scoring was done according to the methods reported by LeMay, et al,
17 see the accompanying table. The motor function of rats was assessed using the Tarlov scale18Neurological Examination
Score Degree of Neurologic Deficit
Consciousness (maximum score 10)
0 normal (no deficit)
2 conscious continuously
4 conscious intermittently
6 stuporous
8 light coma
10 deep coma
Motor Function (maximum score 5)
0 No movement in hind limb,
no weight bearing
1 Barely perceptible movements of
hind limb but no weight bearing
2 Frequent and/or vigorous movement
in hind limb but no weight bearing
3 Can support weight on hind
limb, may take one or two steps
4 Walks with only mild deficit
5 Normal walking
Light Microscopy: Following perfusion the brain was removed from the skull and post fixed by immersion in the same fixative for at least two days before histological processing. Parietal cortex was dehydrated and embedded in paraffin. A series of 10 micrometers thick were cut and stained with cresyl violate.
Cell counting:
A single investigator unaware to research condition assessed three coronal sections per animal obtained at 120 um intervals through the parietal cortex in each experimental group. The number of all neural cells, including those in which the nucleus could not be recognized, and the number of neurons with distinct cytoplasmic and nuclear outlines were counted in four squares with an area of 2500 ěm2, using analysis imaging software (Soft Imaging System) at a total magnification of 400x. Neurons touching the bottom or right borders were included and those touching the upper or left borders were rejected.Statistical Analyses:
Statistical analyses were performed with SPSS for Windows. Data were analyzed using analysis of variance (ANOVA). Statistical significance was assigned to probability values <0.05.RESULTS
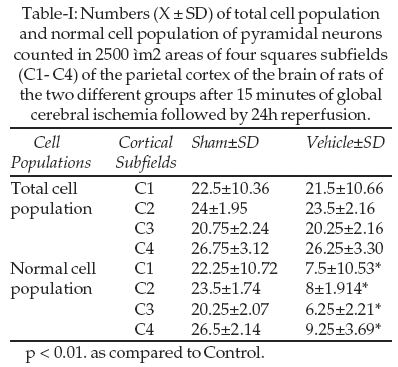
Cell count: Interruption of blood flow to the brain during 15 min through the four-vessel occlusion model followed by 24h reperfusion resulted in a clear reduction of the normal pyramidal neuron population of the cortical subfields of the brain as compared to sham control (Table-I, Figure-1).
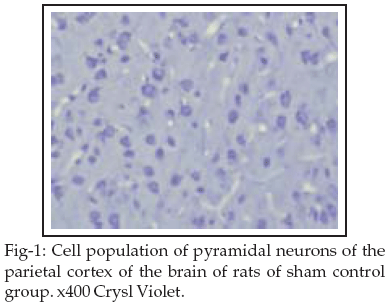
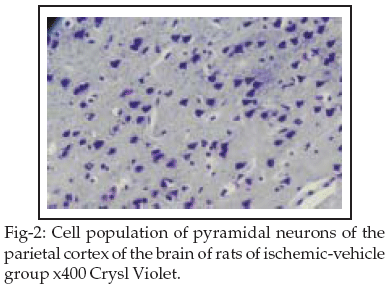
The main loss of pyramidal neurons was observed statistically significant in the C2 subfield of G2 and G3 groups (p < 0.05) as compared to the Vehicle group see Table-II and Figures 2&3.
In addition, brains of these ischemic, diazoxide-treated rats showed a significant increase of the normal cell population of pyramidal cells (p < 0.05) in a dose dependent manner, as compared to the Vehicle group (Table-II Figure-4).
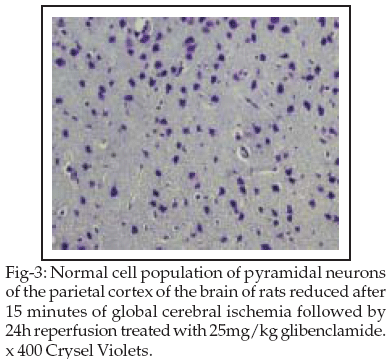
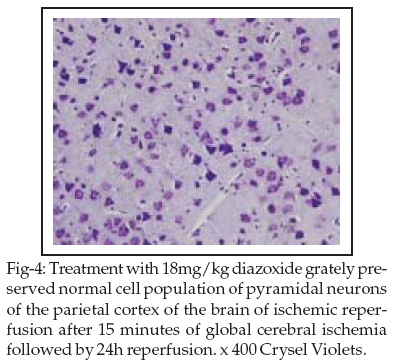
Treatment with glibenclamid prevented the loss of neurons in the C2 subfields of G2 group and both subfields of C1 and C2 of G3 group following the Ischemic/Reperfusion as compared to the Vehicle group. No effect on the neuronal loss was found in diazoxide treated rats in relation to vehicle-treated rats (Table-II).
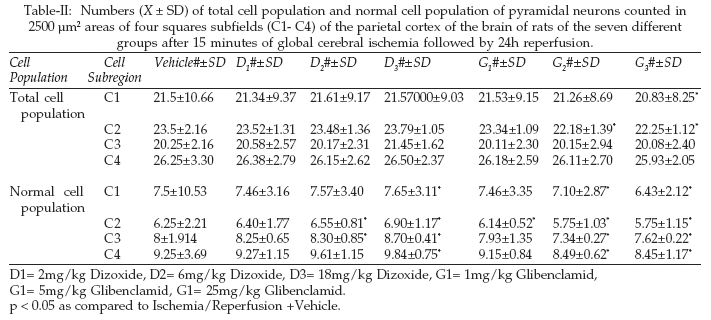
Diazoxide-treated rats showed significant increase in normal cell population of pyramidal neurons of the C2 and C3 subfields of D2 group of rats and C1-C4 subfields of D3 group of rats as compared to Vehicle-treated rats. Glibenclamid significantly reduced normal cell population of pyramidal neurons in C2 subfield of G1 group of rats. Animals in the G2 and G3 groups had significant neuronal damage in all cortex subfields compared with the Vehicle group of rats. Low dose of the glibenclamid (G1 group),
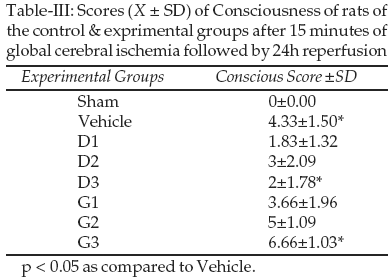
however, had no reduced neuronal counts in the cortex, except for the C2 subfield. The most pronounced damage of all examined cortex subfields was found in the C2 region, which was to be more vulnerable to ischemia/reperfusion.Neurological Deficit: Our result showed that consciousness for vehicle group reduced significantly compared to sham control. ATP dependent potassium channel regulators, diazoxide and glibenclamide significantly affect consciousness following ischemia reperfusion. Average Conscious Scores for the D3 group was 2±1.78, compared to 4.33±1.50 for the Vehicle group and 6.66±1.03 for the G3 group Table-III.
There were no significant differences among animals treated with low dose of diazoxide and glibenclamid compared to Vehicle group. Four artery occlusion significantly reduced motor function compared to sham control. ATP dependent potassium channel regulators, glibenclamid significantly affect motor function following ischemia reperfusion (Table-IV). There were no significant differences among animals treated with Diazoxide compared to Vehicle group.

DISCUSSION
Mitochondrial ATP-sensitive potassium MAK channels play a key role in modulating neuronal survival under ischemic conditions. Diazoxide is a MAK channel opener that has been shown to attenuate the response to ischemia reperfusion injury.
16 It is claimed that MAK channel activation mediates preconditioning.19,20 However the precise mechanisms of diazoxide-induced neuroprotection are largely unexplored at this time. Our present study investigated the effect of diazoxide in an in vivo model of neuronal ischemia reperfusion and the possible mechanisms of diazoxide-induced neuroprotection.It was reported that administration of diazoxide to newborn piglets enhances functional recovery after transient global cerebral ischemia.21 Thus, activation of MAK channels may protect neurons against different kinds of ischemic insults. The mechanism by which activation of these channels is being translated into the observed protection is not known.Several observations suggest that the protective effects of diazoxide in the present study were not due to a vascular action of this drug. Previous studies have shown that effects of diazoxide on blood pressure and flow are due to activation of plasma-membrane potassium channels.
8 Furthermore, the neuroprotective actions of diazoxide in vivo were largely abolished by glibenclamide, strongly suggesting that the effects of diazoxide were mediated by activation of MAK channels. In addition, a previous study indicated that opening of these channels is involved in the regulation of cell viability.7 In agreement with these results we demonstrated the effect of diazoxide on neuronal cell count.Our findings suggest that diazoxide that activate MAK are candidates for use in clinical trials in human stroke patients. Diazoxide has been used in the clinical setting for more than 30 years, primarily for the treatment of patients with acute and severe hypertension.
The direct neuroprotective action of diaz- oxide, suggest that this drug may be particularly effective in preventing neuronal damage after a stroke. However, for diazoxide to be beneficial in human stroke patients, it must be effective when given within a defined post ischemic period, which remains to be established.
ACKNOWLEDGEMENTS
We would like to thank anatomy, physiology, cellular and molecular center of Iran University of Medical Sciences for their help and assistance in this study.
REFERENCES
1. Nagahiro S, Uno M, Sato K, Goto S, Morioka M, Ushio Y. Pathophysiology and treatment of cerebral ischemia. J Med Invest 1998;45:57-70.
2. Wolf PA, Kannel WB, D’Agostino RB. Epidemiology of stroke. In: Cerebrovascular disease: pathophysiology, diagnosis, and management. Eds. MD Ginsberg and J Bogousslavsky. Blackwell Science, Malden, Massachusetts. 1998;834-49.
3. Goldstein LB. Basic and clinical studies of pharmacologic effects on recovery from brain injury. J Neural Transplant Plast 1993;4:175-192.
4. Mattson MP, Clumsee C, Yu ZF. Apoptotic and antiapoptotic mechanisms in stroke. Cell Tissue Res 2000;301:173-87.
5. Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev 2000;80:315-60. A. Saraste and K. Pulkki. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc Res 2000;45:528-37.
6. Holmuhamedov EL, Jovanovic S, Dzeja PP, Jovanovic A, Terzic A. Mitochondrial ATP-sensitive K+ channels modulate cardiac mitochondrial function. Am J Physiol 1998;275:1567-76.
7. Garlid KD. Cation transport in mitochondria. Biochim Biophys Actan 1996;1275:123-6. Halestrap AP. Regulation of mitochondrial metabolism through changes in matrix volume. Biochem Soc Trans 1994;22:522-9.
8. Szewczyk A, Czyz A, Wojcik G, Wojczak L, Nalecz M. ATP-regulated K+ channel in mitochondria: pharmacology and function. J Bioenerg Biomembr 1996;28:147-152.
9. L Wang G, Cherednichenko, L. Hernandez. Preconditioning limits mitochondrial Ca2+ during ischemia in rat hearts: role of KATP channels. Am J Physiol Heart Circ Physiol 2001;280.
10. Szewczyk A, Czyz A, Wojcik G, Wojczak L, Nalecz M. ATP-regulated K+ channel in mitochondria: pharmacology and function. J Bioenerg Biomembr. 1996;28:147–152 McPherson, BC, Yao Z. Morphine mimics preconditioning via free radical signals and mitochondrial KATP channels in myocytes. Circulation 2001;103:290-5.
11. MichaelVC, Baines CP, Downey JM. Ischemia preconditioning: from adenosine receptor to KATP channel. Annu Rev Physiol 2000;62:79-109.
12. Olivetti G, Quaini F, Sala R, Lagrasta C, Corradi D, Bonacina E, et al. Acute myocardial infarction in humans is associated with activation of programmed myocyte cell death in the surviving portion of the heart. J Mol Cell Cardiol 1996;28:2005-16.
13. Garlid KD, Paucek P, Yarov-Yarovoy V, Murray HN, Darbenzio RB, D’Alonzo AJ, et al. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels. Possible mechanism of cardioprotection. Circ Res 1997;81:1072-82.
14. LeMay DR, Gehua L, Zelenock GB. Insulin Administration Protects Neurologic Function in Cerebral Ischemia in Rats. Stroke 1988;19:1411-19.
15. Tarlov IM, Klinger H. Spinal cord compression studies. Arch Neurol Psychiat 1954;71:271-90.
16. Bela K, Nishadi C, Rajapakse, James A, Snipes, Krisztina Nagy, Takashi Horiguchi, David W. Busija. Diazoxide induces delayed pre-conditioning in cultured rat cortical neurons. J Neurochemistry 2003;87:969 .
17. LeMay DR, Gehua L, Zelenock GB, et al. Insulin Administration Protects Neurologic Function in Cerebral Ischemia in Rats. Stroke 1988;19:1411-9.
18. Tarlov IM, Klinger H. Spinal cord compression studies. II: Time limits for recovery after acute compression in dogs. AMA Arch Neurol Psychiatry 1954:71:271-90.
19. Domoki F, Perciaccante JV, Veltkamp R, Bari F, Busija DW, Mitochondrial potassium channel opener diazoxide preserves neuronal – vascular function after cerebral ischemia in newborn pigs. Stroke 1999;30:2713-8.
20. Richer C, Pratz J, Mulder P, Mondot S, Giudicelli JF, Cavero I, Cardiovascular and biological effects of K+ channel openers, a class of drugs with vasorelaxant and cardioprotective properties. Life Sci 1990;47:1693-705.
21. Breslin DJ. Hypertensive crisis. Med Clin North Am 1996;53(2):351-60.
HOME | SEARCH | CURRENT ISSUE | PAST ISSUES
Professional
Medical Publications
Room No. 522, 5th Floor, Panorama Centre
Building No. 2, P.O. Box 8766, Saddar, Karachi - Pakistan.
Phones : 5688791, 5689285 Fax : 5689860
pjms@pjms.com.pk