|
|
||||
|
Published by : PROFESSIONAL MEDICAL PUBLICATIONS |
||||
|
ISSN 1681-715X |
||||
|
||||
|
- |
||||
|
REVIEW ARTICLE |
||||
|
- |
||||
|
Volume 23 |
October - December 2007 (Part-I) |
Number 5 |
||
|
|
||||
|
|
||||
|
|
||||
|
Published by : PROFESSIONAL MEDICAL PUBLICATIONS |
||||
|
ISSN 1681-715X |
||||
|
||||
|
- |
||||
|
REVIEW ARTICLE |
||||
|
- |
||||
|
Volume 23 |
October - December 2007 (Part-I) |
Number 5 |
||
|
|
||||
|
|
||||
Adult onset still’s disease and prurigo pigmentosa:
An unusual association and review of literature
Tarek Abdel Hamed Mostafa Dowod1, M. El-Sayed
Hanafy Khalel2, Magdy Abas Ali Mohammed3,
Maha Abdel Wahab Mohamed El Sharawy4, Sara Jassim Al-Ghabandi5,
Anwaar Yahya Al-Sumait6
SUMMARY
This study was conducted in the Department of Medicine, Department of Dermatology, Al-Adan Hospital, Ministry of Health- Kuwait. This patient was hospitalized as a case of pyrexia of unknown origin. We report atypical skin eruption in this patient who fulfilled the criteria for Adult Onset Still’s Disease (AOSD) despite the absence of the typical still’s rash. Skin biopsy revealed prurigo pigmentosa (PP). To elucidate this unusual association, we searched the literature for the cutaneous manifestations of AOSD. Kahori T, et al had described PP like lesions in a patient with AOSD in addition to the typical still’s rash and it is the only case report that exists in English literature. In our case, except for still’s rash, all the criteria of AOSD were present, the diagnosis was made after excluding all other possible causes i.e. infectious diseases, other rheumatic diseases, vasculitis and malignant disease. The objective for reporting this case and review of literature is to shed more light on the rare dermatological disorder Prurigo Pigmentosa (PP) and its unusual association with Adult Onset Still’s Disease (AOSD).
Clinicians and dermatologist must be aware of this unusual association of PP and AOSD as the diagnosis of AOSD can be made in the absence of the typical still’s rash but in the presence of other atypical cutaneous manifestations including PP.
KEY WORDS: Prurigo pigmentosa, Adult Onset Still’s Disease, Still’s rash.
Abbreviations: Adult onset still’s disease (AOSD). Prurigo Pigmentosa (PP).
Pak J Med Sci October - December 2007 (Part-I) Vol. 23 No. 5 792-797
1. Tarek Abdel Hamed Mostafa Dowod,
Specialist, Dept. of Medicine,
2. Mohammed El-Sayed Hanafy Khalel,
Specialist, Dept. of Dermatology,
3. Magdy Abas Ali Mohammed,
Registrar, Dept. of Medicine,
4. Maha Abdel Wahab Mohamed El Sharawy,
Lecture of Pathology, Dept. of Pathology,
Ain Shames University, EGYPT.
5. Sara Jassim Al-Ghabandi,
Trainee, Dept. of Medicine,
6. Anwaar Yahya Al-Sumait
Trainee, Dept. of Medicine,
1-3,5,6: Al-Adan Hospital, Kuwait.
Correspondence
Dr. Tarek Abdel Hamed Mostafa Dowod,
Al-Adan Hospital, Ministry of Health,
Fahaheel, P.O Box: 47854, Kuwait.
E-mail: doriya70@hotmail.com
* Received for Publication: August 9, 2007
* Accepted: September 15, 2007
INTRODUCTION
Both prurigo pigmentosa (PP) and adult onset still’s disease (AOSD) independently are uncommon diseases. Expectedly the association of PP and AOSD is extremely rare.
PP is a rare dermatosis with unknown etiology.
1 It is seen most commonly among young adult Japanese females.2 Clinically, it presents itself as pruritic urticarial papules, papulo-vesicular and vesicles arranged in a reticulate pattern and distributed symmetrically on the face, neck, back, chest and extremities.3 Lesions involute in a matter of days leaving behind net like pigmentation. Exacerbations and recurrences are the rule.4AOSD is an inflammatory disorder used to describe a series of adult patients who did not fulfill criteria for classic rheumatoid arthritis but had features similar to children with systemic rheumatoid arthritis.
5,6 To establish a diagnosis of AOSD requires the presence of certain major or minor criteria or a combination of both, and absence of certain exclusions.7Prurigo pigmentosa has been described in association with ketosis related to diabetes mellitus, anorexia nervosa and strict diet,
8 pregnancy,9 primary biliary cirrhosis and Sjorgen syndrome.10 Kahori T, et al described PP like lesions in a patient with AOSD in addition to the typical still’s rash and it is the only case report that exists in English literature.11We report a second case of prurigo pigmentosa in association with adult onset still’s disease in absence of the typical still’s rash. As the clinicians and most of the dermatologist are unaware of this unusual association of PP and AOSD, we decided to report this case and to shed more light on both conditions.
CASE REPORT
A 26 - year old Nepali female, a mother of one child with no significant past medical history presented with a ten-day history of daily spiking fever, chills, arthralgias, myalgias and pruritic skin eruption.
On examination, she looked pale, febrile (temperature 39.2şC), had non-suppurative pharyngitis and generalized lymphadenopathy. The lymph nodes were slightly tender, small and discrete. The cardio-respiratory, abdominal and neurological systems were normal. There was no evidence of active arthritis. The skin eruption was in the form of pruritic papular eruption arranged in a reticulate pattern and distributed symmetrically on the face, neck, trunk and legs (Figs-1a, 1b, 1c).
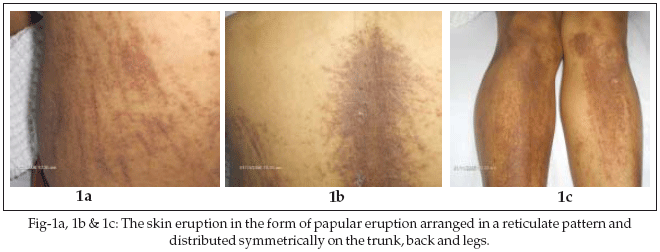
Investigations: CBC: WBC 13.95 X 10
9/l, differential count showed neutrophilic leukocytosis (11.65 X 109/l), Hb 10.1g/dl, MCV 82.3 fl (79.4-94.8 fl), MCHC 26.4 pg (25.6-32.2 pg), platelet 314 X 109/l (182-369 109/l), ESR 115mm/hour, C-reactive protein 201ng/l, ASO titer < 200 IU/ml), liver function tests: AST 131 IU/l (0-31), ALT 273 IU/l (0-35), ALP 266 (35-104 IU/l), total bilirubin 15.9µmol/l (3-22). S. iron 8.25µmol (6.6-26) and S. transferrin 3.91g/l (2.52-4.29) were normal. S. ferritin 2000ng/ml (10-120) was very high.Coagulation profile was normal. Other biochemical profile including renal profile, lipid profile, S. calcium, S. magnesium, phosphorus were within normal limits. S. pregnancy test was negative. Complete sepsis work-up including throat swab, urine routine, blood culture, thick and thin blood film for malaria, Widal test, Brucella agglutination tests, monospot test, CMV serology, mycoplasma serology, HIV serology, hepatitis serology were all negative. Collagen screen (RA factor, ANA, ANCA, anti-mitochondrial antibody, thyroid autoantibodies) was negative. Screening for occult malignancy (stool for occult blood, tumor markers) was negative.
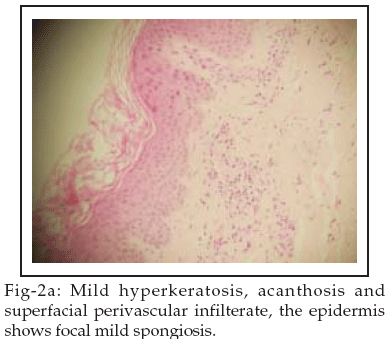
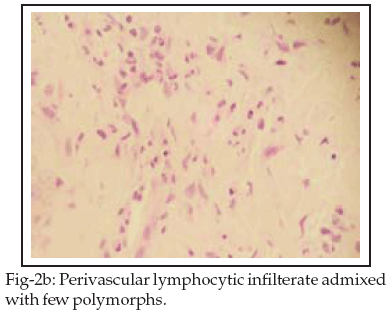
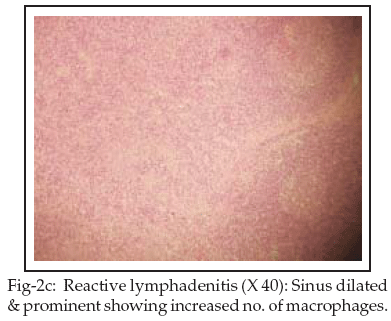
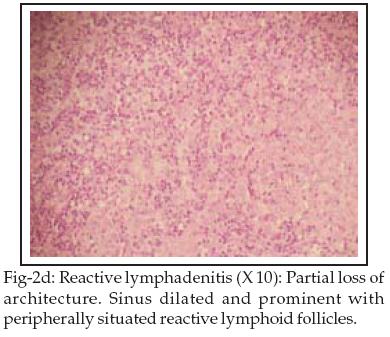
Chest X-ray, ECG, abdominal ultrasound, echocardiography, intravenous urogram did not reveal any abnormalities. CT scan chest, abdomen and pelvis revealed discrete axillary, para-vascular, aorto-pulmonary, left para-aortic lymphadenopathy. Cervical lymph node biopsy revealed reactive lymphadenitis. Bone marrow examination was consistent with anemia of chronic illness with no evidence of hematological malignancies or lymphomatous infiltration. Skin biopsy was consistent with PP (Figs-2a, 2b, 2c, 2d).
MANAGEMENT
The patient was initially managed conservatively with adequate hydration, acetaminophen tablet, as required. However, she complained of daily spiking fever up to 41şC associated with chills and sweating.
The diagnosis of adult-onset Still’s disease was made after excluding the infective, collagen and malignant disorders, supported by the markedly elevated serum ferritin level. The eruption and fever subsided after the administration of prednisolone 1mg/kg/day. It required almost a week for fever to resolve completely. The patient was discharged on a steroid tapering dose. On follow-up, ESR, C-reactive protein and S. ferritin level returned to levels within the normal reference range.
DISCUSSION
The diagnosis of AOSD can be very difficult. There are no specific tests and reliance is usually placed on a symptom complex and the well described typical rash seen in most patients. The classic evanescent rash of still’s disease was first noted by Boldero in 1933
12 and is referred to as a still’s rash or rheumatoid rash despite the absence of an association with adult sero-positive rheumatoid arthritis.To establish a diagnosis of AOSD requires the presence of certain major or minor criteria or a combination of both, and absence of certain exclusions.
7Major Criteria: The proposed major criteria include:-
* Fever of at least 39şC lasting one week or longer.
* Arthralgias or arthritis lasting 2 weeks or longer.
* Characteristic rash which is a macular or maculo-papular, non pruritic salmon-pink eruption usually apparent over the trunk or extremities during febrile episodes (still’s rash).
* Leukocytosis (10,000 X 10
9/l or greater with 80% or more granulocytes.Minor Criteria:
* Sore throat.
* The recent development of significant lymph node swelling.
* Hepatomegaly or splenomegaly.
* Abnormal liver function studies, particularly aminotransferases and lactate dehydrogenases.
* Negative tests for anti-nuclear antibody and rheumatoid factor.
Exclusions Criteria: The following findings must not be present in AOSD:
* Infections such as infectious mononucleosis or parvovirus B
19.* Malignancy, particularly lymphoma.
* Other rheumatic diseases such as polyarteritis nodosa, systemic lupus erythematosus or rheumatoid arthritis, vasculitis with extra-articular features.
Six sets of criteria have now been proposed for establishing the diagnosis of AOSD.
13-18 These sets are similar with the difference being the number of major and minor criteria required. A comparison of these six sets of criteria demonstrated that the Japanese criteria had the greatest sensitivity in establishing the diagnosis. The Japanese criteria requires the presence of five features with at least two being major diagnostic criteria.While 92% of all patients demonstrate some cutaneous manifestations during their illness, the more specific still’s rash is seen in 86% of patients with AOSD.
19 Other cutaneous manifestations of AOSD have been reported but these are not well known.Cutaneous manifestations of AOSD: The common ones are Still’s Rash, Koebner phenomenon, Dermatographism.
Uncommon: It includes Pruritis, Urticaria, Dermal plaques, Facial Rash, Alopecia, Erythema nodosumd and Raynaud’s phenomenon.
AOSD has been associated with markedly elevated serum ferritin level in as many as 70% of patients with disease activity and has been suggested as a serologic marker to monitor the response of treatment.
20PP was first described by Nagashima, et al
21,22 in 1971 in Japan and is diagnosed most commonly in the Japanese population. Less than 40 non-Japaneses cases have been published.23-27 Clinically, it presents itself as pruritic urticarial papules, papulo-vesicular and vesicles arranged in a reticulate pattern and distributed symmetrically on the face, neck, back, chest and extremities.3 Lesions involute in a matter of days leaving behind net like pigmentation. Exacerbations and recurrences are the rule.4Histopathologically, PP begins with superficial perivascular infilterate of neutrophils, shortly thereafter, neutrophils are scattered in dermal papillae and then sweep rapidly through an epidermis in which spongiosis, ballooning and necrotic keratocytes are accompaniments. Very soon eosinophils and lymphocytes come to predominate over neutrophils in a dermal infilterates that assumes a patchy lichenoid pattern. Intraepidermal vesiculation follows on spongiosis and ballooning and sometimes subepidermal vesiculation on vacuolar alteration at the dermo-epidermal junction. As the epidermis becomes hyperplastic, parakeratotic and slightly hyperpigmented melanophages begin to appear in the dermis. Studies by immunofluroresence are negative invariably.
24Prurigo pigmentosa has been described in association with ketosis related to diabetes mellitus, anorexia nervosa and strict diet,
8 pregnancy,9 primary biliary cirrhosis and Sjorgen syndrome.10 To diagnose PP with surety in a patient, clinico-pathologic correlation is often necessary. Diseases to be considered in the clinical differential diagnosis at an early stage of the process are early stages of dermatitis herpetiformis, linear IGA dermatosis, and acute lupus erythromatosis, even though the trunk-centered distribution of lesions and the reticular arrangement of individual lesions in PP allow differentiation. At a resolving stage of the process, PP may be confused with reticular pigmented papules of confluent and reticulated papillomatosis of Gougerot and Carteaud, but in contrast to those lesions, pigmented macules of PP are not keratotic.Differential Diagnosis:
Diseases that have to be considered in the differential diagnosis histopathologically vary according to the stage of the disease process. Early in the course, urticaria, an early manifestation of leukocytoclastic vasculitis, dermatitis herptiformis, linear IGA dermatosis, acute lupus erythromatosis, eruptive psoriasis, or dermatophytosis may be considered because of the predominance of neutophils in the infilterate, but epidermal changes typical of PP, such as spongiosis combined with ballooning, scattered neutrophils in the epidermis, and or necrotic keratinocytes in the absence of features diagnostic of any of the conditions mentioned usually enable differentiation. A fully developed lesion of PP has to be differentiated from erythema multiforme and Mucha-Habermann disease. Neutrophils in collections beneath the cornfield layer and few eosinophils in the infiltrate are clues to the diagnosis of PP. At a late stage of the process, histopathologic features of PP are indistinguishable from any other disease that resolves with postinfammatory hyperpigmentation. With clinicopathologic correlation, however, the disease may still be diagnosed with certainty because of the typical netlike distribution of pigmentation concerned on the trunk.28Treatment:
Several medications are available for treatment of PP, minocycline and dapsone being used most commonly. Both medications have in common antibiotic and anti-inflammatory effects and they are especially effective in the inhibition of migration of neutrophils, a mechanism that may explain their efficacy in PP. A recent report highlights favorable response of PP to isotretinoin, the mechanism of action of this medication being opaque.27 Many patients also experience spontaneous resolution of the eruption after a few weeks, recurrences occurring only months or years later.Kahori T, et al described PP like lesions in a patient with AOSD in addition to the typical still’s rash and it was the only case report exists in English literature.
11 In this case, the patient developed PP-like lesions in addition to the typical rash of AOSD. In our case, except for still’s rash, all the criteria of AOSD were present, with support from highly raised serum ferritin level, the diagnosis was made after excluding all other possible causes i.e. infectious diseases, other rheumatic diseases, vasculitis and malignant disease.CONCLUSIONS
The diagnosis of AOSD can be made in the absence of the typical still’s rash but in the presence of other atypical cutaneous features. Therefore, we should carefully follow the clinical course of a patient in order not to overlook these atypical cutaneous manifestations of ASOD.
ACKNOWLEDGMENT
The authors thank Dr. Jaffer Ismail Ali for his kind cooperation and support in the preparation of this manuscript.
REFERENCES
1. Liu MT, Wong CK. Prurigo pigmentosa. Dermatology 1994;188:219-21.
2. Gur-Toy G, Gungor E, Artuz F, Aksoy F, Alli N. Prurigo-pigmnetosa. Int J Dermatol 2002;41:288-91.
3. Boer A, Misago N, Walter M, Kiryu H, Wang XD, Ackerman AB. Prurigo pigmentosa: a distinctive inflammatory disease of the skin. Am J Dermatopathol 2003;25:117-29.
4. Matsumoto C, Kinoshita M, Baba S, Suzuki H, Kanematsu S, Kanematsu N. Vesicular Prurigo pigmentosa cured by minocycline. J Eur Acad Dermatol Venereol 2001;15:354-6.
5. Still GF. On a form of chronic joint disease in children. Med Chir Trans 1897; 80:47. (Reprinted in: Arch Dis Child 1941;16:156).
6. Bywaters EG. Still’s disease in the adult. Ann Rheum Dis 1971;30:121.
7. Masson C, LeLoet X, Liote F. Comparative study of six types of criteria in adult still’s disease. J Rheumatol 1996;23:495.
8. Nakada T, Sueki H, Lijima M. Prurigo pigmentosa (Nagashima) associated with anorexia nervosa. Clin Exp Dermatol 1998;23:25-7.
9. Yokozeki M, Watanabe J, Hotsubo T, Matsumura T. Prurigo pigmentosa disappeared following improvement of diabetic ketosis by insulin. J Dermatol 2003;30:257-8.
10. Togawa Y, Shinkai H, Utani A. Prurigo Pigmentosa in a patient with primary biliary cirrhosis and Sjogren syndrome. J Dermatol 2004;31:815-9.
11. Kahori T, Yayoi N, Naoko O, Michiko H, Yukie E, Atushi T, et al. Adult-onset still’s disease with prurigo pigmentosa-like skin eruption. J Dermatology 2006;33:55-8.
12. Boldero MEA. A case of still’s disease. Mod Soc Trans London 1933;16:55.
13. Yamaguchi M, Ohta A, Tsunematsu T. Preliminary criteria for classification of adult still’s disease. J Rheumatol 1992;19:424.
14. Calabro JJ, Marchesano JM. Fever associated with juvenile rheumatoid arthritis. N Engl J Med 1967;276:11.
15. Reginato J, Schumacher HR Jr, Baker DG. Adult onset still’s disease. Experience in 23 patients and literature review with emphasis on organ failure. Semin Arthritis Rheum 1987;17:39.
16. Cush JJ, Medsger TA Jr, Christy WC. Adult-onset still’s disease. Clinical course and outcome. Arthritis Rheum 1987;30:186.
17. Goldman JA, Beard MR, Casey HL. Acute febrile juvenile rheumatoid arthritis in adults. Causes of polyarthritis and fever. South Med J 1980;73:555.
18. Kahn MF, Delaire M, Maladie de still de l’adulte. In: Kahn MF, et al. eds, Les maladies systemiques. Paris 1991; Flammarion.
19. Kaur S, Bamberry P, Dhar S. Persistent dermal plaque lesions in adult-onset still’s disease. Dermatology 1994;188:241-2.
20. Lambotte O, Cacoub P, Costedot N. High ferritin and low glycosylated ferritin may also be a marker of excessive macrophage activation. J Rheumatol 2003;30:1027.
21. Nagashima M, Ohshiro A, Shimizu N. A peculiar pruginious dermatosis with gross reticular pigmentation [in Japanese]. Jpn J Dermatol 1971;81:38-9.
22. Nagashima M. Prurigo pigmentosa-clinical observations of our 14 cases. J Dermatol 1978;5:61-7.
23. Boer A, Misago M, Wolter M, Kiryu H,Wang XD, Ackerman AB. Prurigo pigmentosa: new observations and comprehensive review. Avaliable from: URL:http://www.derm101.com. Accessed 2006.
24. Boer A, Misago N, Wolter M, Kiryu H, Wang XD, Ackerman AB. Prurigo pigmentosa: a distinctive inflammatory disease of the skin. Am J Dermatopathol 2003;25:117-29.
25. Boer A, Ackerman AB. Prurigo pigmentosa (Nagashima disease), textbook and atlas of a distinctive inflammatory disease of the skin. New York: Ardor Scribendi Ltd; 2004.
26. Daneshpazhooh M, Safar F, Barikbin B, Safee-naraghi Z, Asgari M, Chams- Davatchi C. Vesicular prurigo pigmentosa in a 16-year old girl. Nouv Dermatol 2004;23:599-603.
27. Requena Caballero C, Nagore E, Sanmartin O, Botella- Estrada R, Serra C, Guillen C. Vesiculous prurigo pigmentosa in a 13- year-old girl: Good response to isotretinoin. J Eur Acad Dermatol Venereol 2005;19:474-6.
28. Masoud A, Maryam D, Cheyda CD, Boer A. Prurigo pigmentosa: An underdiagnosed disease in patients of Iranian descent? J Am Acad Dermatol 2006;55:131-6.
HOME | SEARCH | CURRENT ISSUE | PAST ISSUES
Professional
Medical Publications
Room No. 522, 5th Floor, Panorama Centre
Building No. 2, P.O. Box 8766, Saddar, Karachi - Pakistan.
Phones : 5688791, 5689285 Fax : 5689860
pjms@pjms.com.pk